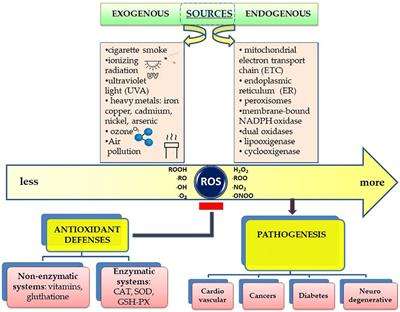
Cancer is the second cause of death worldwide and is characterized by several hallmarks47; cell transformation, genome instability, hyperproliferation, immortalization, angiogenesis, epithelial-mesenchymal transition (EMT) and metastasis, which are all influenced in several ways by intracellular ROS48,49.
ROS as double-edged swords in cancer
Several noncancer cells associate with tumors: among these, cancer-associated fibroblasts (CAFs), particularly represented in the tumor microenvironment (TME), actively contribute to the regulation of tumor homeostasis, promoting tumor progression and the invasion of cancer cells. CAFs and ROS engage in two-way cross-talk: on the one hand, fibroblasts are targeted by ROS, particularly H2O2, which is able to convert them into active CAFs through the upregulation of HIF1α; on the other hand, CAFs are critical for the increase in ROS levels observed in cancer50,51. CAFs can also promote cancer growth and invasiveness, and both CAFS and ROS are linked through the increases in ROS-generated CAFs to which most cancers respond by increasing the expression of antioxidant genes52,53,54 (Fig. 2).
Fig. 2: The three types of programmed cell death induced by elevated ROS levels in cancer cells.
ROS, in response to death-inducing ligands (TNFα and Fas), enhance the assembly of DISCs and the activation of effector caspases and reduce Bcl-2 activity or, as a consequence of increased permeability of mitochondrial PTPs, stimulate the intracytoplasmic release of cytochrome c, which interacts with Apaf-1 and procaspases and forms the apoptosome (apoptosis). ROS can also inhibit the negative regulators of autophagy (TORC1) and increase the formation of LC3-dependent autophagosomes (autophagy). Finally, high levels of ROS, induced by several receptor-interacting protein kinases (RIPs), increase p53 expression, which increases ROS levels via a mechanism that depends on intracellular iron (ferroptosis).
Full size image
However, a growing body of evidence supports the view that antioxidant activities are essential for tumorigenesis. It has been recently reported that targets of the Nrf2 gene, such as HMOX1, facilitate cancer development because they counteract the effect of oxidative stress in transformed cells55. Moreover, established oncogenes such as K-RAS and c-MYC, which had been previously demonstrated to induce intracellular ROS56,57, have been recently shown to stabilize Nrf258. In this regard, mutations to NRF2 and its regulator KEAP1 have been found in cancer cells, supporting the supposition that antioxidant genes are pivotal in tumor progression59,60,61,62. In fact, it has been found that the breast cancer susceptibility 1 (BRCA1) gene interacts with and induces Nrf2 expression with positive outcomes on cancer cell survival63. Interestingly, estrogen stimulation of breast cancer cells that do not express BRCA1 and, as a result, suffer from high intracellular ROS levels rescues NRF2 transcription, enhancing the survival of these cancer cells64.
Additionally, FOXO transcription factors have recently been implicated in tumorigenesis: in fact, rhabdomyosarcomas present FOXO genes with a high percentage of mutations that render them insensitive to inhibition by AKT signaling65. Moreover, increased intracellular levels of GSH are required for the initiation and progression of various types of cancer, and inhibitors of GR behave as anticancer drugs66, while high levels of NADPH boost the metastatic ability of melanoma cells, and protocols based on depletion of GSH (isothiocyanates and aziridine derivatives that bind GSH) or based on blocking the uptake of a rate-limiting precursor of its synthesis (inhibitors of the cysteine/glutamate antiporter, XCT) greatly impact cancer cell survival67,68. Specifically, sulfasalazine, an XCT inhibitor, appears useful in the treatment of pancreatic and small-cell lung cancer cells69,70, while NOV-002, a glutathione disulfide mimetic that alters the GSSG/GSH ratio and induces oxidative stress, has been favorably used in patients with HER2-negative breast cancer71. In addition, inhibitors of the enzyme glutaminase (GLS) that converts glutamine to glutamate, which is subsequently transformed to GSH via the glutamate–cysteine ligase complex, efficiently induce cancer cell death through dysregulation of their antioxidant system72. As mentioned above, another central player in these redox systems is thioredoxin, which is reduced by NADPH to induce the transfer of electrons for use in DNA synthesis, signal transduction and redox regulation. Interestingly, auranofin, which functions as a thioredoxin inhibitor, has been used with beneficial effects in the treatment of head and neck carcinoma cell lines; prevention of this effect by the ROS scavenger N-acetylcysteine (NAC) confirms the role of ROS in these cancers73.
ROS and apoptosis (type I programmed cell death)
The most common method by which ROS kill transformed cells is the activation of PCD, which is completed within less than 60 min by a family of cysteine-dependent aspartate-directed proteases known as caspases. Triggered by an extrinsic or an intrinsic pathway, caspase-induced PCD culminates with the formation of apoptotic bodies that are eliminated by adjacent phagocytes74. The extrinsic pathway is mediated by binding of death-inducing ligands such as TNFα and Fas ligand that bind to cognate receptors that, in turn, recruit adaptor proteins and pro-caspases, leading to the assembly of the death-inducing signaling complex (DISC) and the activation of effector caspases75. This interaction is competed by the cellular FLICE-inhibitory protein (c-FLIP): ROS have been shown to downregulate the c-FLIP half-life by inducing its ubiquitin-proteasomal degradation, thus enhancing this extrinsic pathway76. However, compelling evidence suggests that, for the majority of ROS-related anticancer drugs, apoptosis depends on the activation of the intrinsic pathway that involves mitochondrial PTPs, the permeability of which is increased with the cytoplasmic release of pro-apoptotic factors such as cytochrome c that forms a complex with apoptotic protease activating factor 1 (Apaf-1) and pro-caspase 9 to build the apoptosome, activating, in turn, effector caspases77,78,79,80 (Fig. 2).
In fact, ROS induce the three major components critical for the opening of the PTPs, the voltage-dependent anion-selective channel (VDAC), adenine nucleotide translocase (ANT) and cyclophilin D, via the oxidation of specific cysteines in their active sites81,82. ROS also trigger apoptosis by inactivating or increasing the ubiquitination of the pivotal anti-apoptotic protein Bcl-2 and by decreasing the intracellular levels of Bax and Bad83,84 (Fig. 2).
The induction of apoptosis by elevated ROS levels has been highlighted as the central mechanism responsible for the positive effects of monoclonal antibodies85 and tyrosine kinase inhibitors86, which represent the core of targeted cancer therapy87. Among tyrosine kinase inhibitors, imatinib (a PDGFR inhibitor) and erlotinib (an EGFR inhibitor) induce ROS-dependent apoptosis in melanoma and non-small-cell lung cancer cells, respectively, through disruption of mitochondrial membrane potential upon the stimulation of JNK and p38 phosphorylation88,89, while vemurafenib (a BRAF inhibitor) increases the production of superoxide anions with the commensurate depolarization of the mitochondrial membranes in melanoma cells90. Among the monoclonal antibodies, rituximab (specific to the calcium-channel protein CD20 on the surface of B cells and mature plasma cells) increases ROS and induces apoptosis via the inhibition of Bcl-2 and p38MAPK signaling and is used in the treatment of B cell lymphomas91.
As noted above, chemotherapy and radiotherapy cause an increase in intracellular ROS that can lead to apoptosis92,93 via extrinsic or intrinsic pathways94,95. Many drugs used in anticancer therapy induce oxidative stress. Apoptosis is stimulated by procarbazine, which induces oxidative DNA damage that cannot be repaired by the BER/NER system in Hodgkin’s lymphoma and brain cancers96. Doxorubicin-dependent cytotoxicity is linked to the stimulation of a Fenton reaction that generates hydroxyl radicals successfully used for the treatment of Kaposi’s sarcoma, breast and bladder cancer and acute lymphocytic leukemia97. A course of treatment with arabinocytosine, which hampers DNA replication, followed by anthracyclines to increase ROS, has been shown to drive PCD, with beneficial effects for patients with acute myeloid leukemia (AML)98.
Arsenic trioxide has recently carved out a role in cancer therapy because it can induce electron leakage along the respiratory chain99. It triggers apoptosis in different cancer cells, including those of myeloma, lung cancer, and leukemia100,101. Moreover, 5-fluorouracil, a pyrimidine analog, produces ROS through p53-dependent pathways and induces apoptosis in colon and rectal cancer cells102,103.
ROS-induced apoptosis also explains the beneficial effect of two analogs of nuclear receptor ligands in several types of cancer: 2-methoxyestradiol, a 17β-estradiol metabolite, and N-(4-hydroxyphenyl) retinamide, a synthetic analog of retinoic acid, have been shown to induce PCD in neuroblastoma and lung cancer cells, respectively104,105. Furthermore, platinum-based drugs elevate ROS levels that promote PCD; protocols for the administration of these compounds in combination with inhibitors of poly(ADP-ribose) polymerase (PARP), which is involved in the maintenance of DNA integrity, have been shown to arrest the growth of breast cancer cells, even in BRCA-deficient models106,107. Intuitively, the inhibition of DNA damage repair by PARP may sensitize cancer cells to the oxidative stress induced by platinum-containing drugs.
Programmed cell death may also be mediated by the effect of elevated ROS on sphingomyelinase, which generates ceramide from sphingomyelin and binds to death receptors on the cell membrane of cancer cells. Activation of this pathway has been observed after UV irradiation of lymphoma cells108. Moreover, the use of drugs affecting mitochondria, where more than one-half of all ROS are generated, represents a suitable approach to induce oxidative stress and PCD in cancer cells109: gamitrinib, an inhibitor of heat shock protein 90 (HSP90), induces a dramatic collapse of mitochondria in prostate cancer cells110, while ARQ 501 (a quinone derivative) and STA-4783 (a copper chelator) increase ROS through leakage in the electron transport chain and have beneficial effects in patients with solid tumors and pancreatic adenocarcinoma111.
Apoptosis is triggered in cells with excessive endoplasmic reticulum (ER) stress that is induced when the protein folding ability of the ER is overwhelmed or impaired. Recently, several drugs have been designed on the basis of their ability to aggravate ER stress in cancer cells via the induction of oxidative stress. Among these, bortezomib is a proteasome inhibitor that induces ROS and ER stress in head and neck squamous cell carcinoma cells112, and celecoxib, a nonsteroidal anti-inflammatory drug, aggravates ER stress and induces apoptosis by altering the Bax/Bcl-2 ratio and increasing ROS in prostate cancer cells113.
ROS and autophagy (type II programmed cell death)
Recently, an important therapeutic approach to kill cancer cells has been presented by ROS-induced autophagy114. Specifically, it has been reported that H2O2-dependent inactivation of autophagy-related gene-4 (ATG4) increases LC3-associated autophagosomes and that ATM-mediated oxidation of AMP-activated protein kinase (AMPK) inhibits mammalian target of rapamycin 1 (TORC1), a pivotal negative regulator of autophagy115,116,117 (Fig. 2). Indeed, autophagy, also known as type II programmed cell death, is now considered not only as a cell survival mechanism but also a tumor suppressor mechanism that induces the death of transformed cells118. In this regard, it has been reported that H2O2 induces autophagic cell death in glioma cells after treatment with the polycyclic ammonium ion sanguinarine, which increases electron leakage from mitochondria and induces NOXs119. Rapamycin, administered in combination with inhibitors of HSP90, causes mitochondrial damage with accompanying oxidative stress and autophagy and reduces tumor growth in RAS-dependent tumors120.
ROS and necroptosis (type III programmed cell death)
ROS are also able to induce necrosis, which was originally considered an unregulated form of cell death but is now recognized as type III programmed cell death (necroptosis)121,122. ROS generated after the formation of ceramide or after an increase in energy metabolism induced by several receptor-interacting protein kinases (RIPs), either in the mitochondrial ETC and/or by NOXs, have been reported to enhance necroptosis123,124,125.
In addition, a very intriguing ROS-related molecular mechanism of tumor suppression by p53 has recently been highlighted; this protein induces a peculiar form of cell death, now called ferroptosis, via an increase in ROS levels that subsequently inhibit the cystine uptake typically mediated by the repression of a key component of the cystine/glutamate antiporter126. Ferroptosis depends on the presence of intracellular iron and is induced by ROS127 (Fig. 2). Therefore, the role of p53 in this context appears to be different from that reported in several studies showing that it decreases the levels of ROS. A plausible explanation of this apparent dichotomy is that p53 promotes cell survival by preventing excessive increases in ROS under moderate oxidative stress, whereas when the oxygen species increase over a threshold level, it switches to becoming a ROS inducer, triggering cell death. On the basis that ferroptosis is considered an oxidation-induced cell death mechanism, several trials with different drugs that elicit this pathway have been conducted128,129. Erastin is a synthetic drug that induces cell death through ferroptosis in tumor cells bearing mutant RAS by increasing intracellular ROS levels and altering the permeability of the outer mitochondrial membrane130,131.
ROS and multidrug resistance
Increased ROS levels are thought to impair the multidrug resistance of cancer cells, which causes cancer development and metastasis during or after chemotherapy132,133. It has been recently shown that efflux pumps in the plasma membrane of cancer cells are crucial for the extracellular efflux of anticancer drugs134. These pumps belong to the adenosine triphosphate (ATP)-binding cassette (ABC) transporter superfamily and are dependent on intracellular ATP stores135. ATP is accumulated by a synthase driven by a proton gradient generated in mitochondria by the NADH-dependent electron transport chain136,137; therefore, one possible way to overcome efficient efflux in cancer drugs is to inhibit ATP synthesis by promoting NADH conversion to NAD through lipid membrane-coated silica carbon nanoparticles that, under near-infrared laser irradiation, target mitochondria and produce ROS with simultaneous consumption of NADH138.
Nuclear ROS: a Trojan horse that induces DNA damage
A new role of ROS related to transcriptional output has been recently highlighted. It is well known that cells follow a strictly scheduled program for differentiation that is based on an orchestrated sequence of gene expression. Because of spatial constraints, genes must engage in a complex unfolding process to become accessible to the transcriptional machinery, which is triggered through posttranslational modifications at the N-terminal tails of core histones. Together, these modifications, induced by coordinated targeting of transcription factors that is currently referred to as epigenetic marks, conform to a precise code with specific time requirements to control whole gene expression139. We have previously shown that estrogen-induced transcription is triggered by LSD1-catalyzed demethylation of lysine 9 in histone H3 (H3K9), which is activated by the binding of liganded estrogen receptor to the enhancers of target genes140. This event is followed by the generation of ROS from the oxidation of FADH2 as induced by the demethylase, with consequent oxidation of nearby guanines (8-oxo-Gs) and recruitment of DNA repair enzymes (among which is APE1) that cause single-strand breaks in DNA and enable looping between the enhencer/promoter and the polyadenylation sites of the target genes, with productive transcription140,141. Intuitively, generation of ROS in this process must be timely and spatially controlled to prevent excessive damage to the DNA: a recent report, in fact, describes a new role for the originally discovered superoxide dismutase, SOD1, that is recruited to the nucleus in response to specific stimuli142. However, it has also been observed that hormone-induced phosphorylation of serine 10 in H3 histone (H3S10) prevents the rapid remethylation of the preceding lysine, serving as the metronome of the process and giving the DNA damage repair system enough time to eliminate the oxidized nucleotides from nearby DNA143. It has been reported that by inhibiting phosphorylation of serine 10 in this pathway, breast cancer cells simultaneously challenged with estradiol show an overproduction of ROS, with increased oxidation of the DNA that overwhelms the repair apparatus and triggers PCD in a great percentage of these cells144 (Fig. 3).
Fig. 3: Role of nuclear ROS in transcription and DNA damage.
ROS generated during nuclear receptor-induced transcription of target genes by the activity of lysine demethylases on lysine 9 in histone H3 must be controlled to prevent their accumulation. To this end, SOD1 reaches the nuclear space, while phosphorylation of H3S10 inhibits the rapid remethylation of the same lysine. If inhibitors of the H3S10 kinases are introduced as a Trojan horse together with nuclear receptor ligands, remethylation of H3K9 is quick, nuclear ROS accumulate, and unrepaired DNA damage triggers PCD.
Full size image




