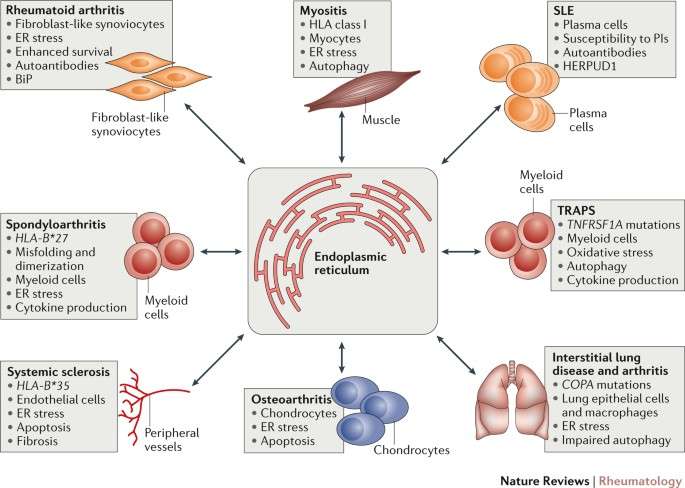
Most of the previous studies, either clinical or preclinical, revealed that Sestrin2 can protect against different cancers and several molecular mechanisms were shown to be involved in such effect.
Chen et al. reported that Sestrin2 has a lower expression in the tissue samples of patients with non-small cell lung cancer, compared with non-cancerous lung tissues [115]. Furthermore, lower expression of Sestrin2 was correlated with poor differentiation of tumor cells, advanced TNM stage, lymph node metastasis, and shorter overall survival [115]. Wei et al. observed that both human colorectal cancer tissues and cell lines have lower expression of Sestrin2, compared with normal colorectal tissue, polyps, and adenomas [18]. Also, lower expression of Sestrin2 was correlated with advanced tumor stage, lymphatic invasion, lymph node metastasis, vascular invasion, and liver metastasis and predicted shorter overall survival and disease-free survival [18]. Similarly, it was reported that Sestrin2 has a lower expression in hepatocellular carcinoma tissue, compared to non-cancerous liver tissue. Meanwhile, Sestrin2 expression decreases in higher stages of the tumor and positively correlates with patients’ survival [116]. Furthermore, lentiviral-mediated upregulation of Sestrin2 has been associated with inhibition of pancreatic cancer cells proliferation, invasion, and migration in PANC-1 and CFPAC-1 cell lines [117].
It was shown that Sestrin2 expression increases in DSS-induced colitis in mice, inhibits mTORC1 and ER stress, and finally facilitates the resolution of inflammation and the healing process [65]. Meanwhile, it was shown that Sestrin2 is controlled by P53 to inhibit mTORC1 and prevent colitis-associated colon cancer [65]. The study indicated that Sestrin2 is downregulated in human colon cancer tissue and SENS2−/− is associated with increased tumor growth and chemoresistance in colitis-associated colon cancer in mice [65]. Consistently, it was shown that activation of the AMPK/mTORC1 pathway by Sestrin2 leads to activation of caspase 3, 7, and 9 and increases apoptotic cell death in colorectal cancer [118]. Interestingly, Wang et al. reported that gastric cancer and colorectal cancer downregulate an E3 ubiquitin ligase RING finger protein 167 (RNF167) and upregulate a deubiquitinase STAMBPL1 that can prevent the ubiquitination of Sestrin2, decrease Sestrin2-GATOR2 interaction and increase mTORC1 signaling [119]. Consistently, knockout of STAMBPL1 and subsequent increase in Sestrin2 ubiquitination markedly inhibited xenograft tumor growth [119].
Importantly, Sestrin2 markedly decreased HIF-1α accumulation in colorectal cancer, even in response to a hypoxic condition or in response to cobalt chloride (CoCl2) as a hypoxia-mimetic agent [57]. Sestrin2 depends on AMPK to enhance the activity of propyl hydroxylase. Propyl hydroxylase converts HIF-1α to OH- HIF-1α and accelerates its degradation [57]. HIF-1α downregulation by Sestrin2 decreased VEGF which is crucial for intra-tumor angiogenesis [57]. HIF-1α downregulation also decreased the expression of glucose transporter 1 (GLUT1) and lactate dehydrogenase A (LAHA) which can impair aerobic glycolysis of tumor cells [57]. Further, Yan et al. indicated that rosemary extract can increase apoptosis and decrease tumor size in the mice model of colon cancer by enhancing the expression of Nrf2 and Sestrin2 [120]. Likewise, Wei et al. revealed that Sestrin2 is downregulated in colorectal cancer and viral vector-mediated upregulation of Sestrin2 reduces colorectal cancer cells proliferation, migration, and colony formation in HCT116 and SW620 cell lines [121]. Viral vector-mediated upregulation of Sestrin2 also decreased tumor size in a mouse xenoplant model [121]. Importantly, they found that Sestrin2 inhibits the Wnt/β-catenin pathway, thereby reducing cancer cells stemness through downregulation of sex-determining region Y-Box 2 (Sox2), octamer-binding transcription factor 4 (Oct4), Kruppel-like factor 4 and c-myc [121]. Cancer stemness crucially potentiates cancer cells self-renewal, promotes their ability to metastasize, maintains their microenvironment, and enhances their potential to resist chemotherapeutic agents [122]. Hence, Sestrin2-mediated attenuation of cancer cell stemness can greatly advance cancer chemotherapy.
Sestrin2, via activation of P38 MAPK, increased the expression of tumor necrosis factor receptor superfamily member 6 (FAS receptor) in A375 and A875 melanoma cell lines and induced their apoptosis [123]. Sestrin2 also enhanced the expression of tumor necrosis factor receptor 1 (TNFR1), related FAS, and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptors to induce apoptosis in the lung adenocarcinoma cells [124]. Sestrin2 stimulated lysosomal degradation of X-linked inhibitor of apoptosis protein (XIAP) to promote receptor-mediated apoptosis of lung adenocarcinoma cells [124]. Similarly, it was shown that Sestrin2 activates apoptosis and mediates the anti-tumor effects of fisetin on human head and neck cancer cells. It suppressed the mTORC1/myeloid cell leukemia 1(Mcl-1) pathway to promote apoptosis in head and neck cancer cells [125]. Mcl-1 is a Bcl-2 family protein and exerts anti-apoptotic effects, hence, Sestrin2 can facilitate apoptosis by downregulating it [126]. Furthermore, SESN2 knockdown strongly reversed fisetin-induced apoptosis in cancer cells [125]. Hence, Sestrin2 is a strong regulator of apoptosis in cancer cells and augments death signals, and downregulates negative regulators of apoptosis in cancer cells.
It was shown that prostate cancer cell lines such as PC3, LNCaP clone FGC, and DU145 have lower expression of Sestrin2, compared with normal prostate epithelial cells [127]. Further, higher expression of Sestrin2 decreased cancer cells proliferation and enhanced their sensitivity to ionizing radiation [127]. Likewise, it was observed that Sestrin2 has a lower expression in bladder cancer, compared with normal neighboring tissue [93] and increased expression of Sestrin2 can inhibit human bladder cancer cells growth [92, 93].
Likewise, overexpression of Sestrin2 was associated with the downregulation of mTORC1 and HIF-1α and increased autophagic flux and apoptosis in osteosarcoma cancer cells [94]. Activation of the Sestrin2/LKB1/AMPK axis attenuated the deleterious effects of mTORC1 signaling and increased nasopharyngeal cancer cells death [89]. Likewise, overexpression of Sestrin2, in an AMPK-dependent manner, improved radiosensitization of breast cancer and colon cancer cells and these effects were abrogated after SESN2 silencing [128, 129]. Shin et al. reported that both Sestrin2 and mTORC1 have higher expression in human endometrial cancer, compared with adjacent normal tissue. Meanwhile, higher expression of Sestrin2 predicted significantly shorter overall and disease-free survival of patients in this study [130]. Interestingly, it was shown that Setrin2 overexpression is significantly correlated with the overactivation of the mTORC1/P70S6K/S6 pathway in human endometrial cancer [130]. Indeed, Sestrin2 overexpression is a compensatory mechanism that attempts to inhibit the mTORC1/P70S6K/S6 pathway in cancer cells and prevent the positive effects of mTORC1 on cancer cells proliferation. Furthermore, Sestrin2, in a mTORC1-dependent manner, inhibited the production of reactive oxygen species (ROS) in cancer cells [130]. Similar to mTORC1 inhibition by rapamycin, lentiviral overexpression of Sestrin2 decreased tumor cells proliferation, EMT, and migration [130]. Also, knockdown of SENS2 inversely promoted the mTORC1 pathway and led to tumor cells proliferation and migration in this study [130]. These findings reveal that Sestrin2 upregulation can be a compensatory response to attenuate the detrimental effect of several signaling pathways and contributes to overcoming resistance to radiotherapy or chemotherapy.
Previously, it was mentioned that Sestrin2 can attenuate inflammasome activation. Attenuation of inflammasome response by activating the Sestrin2/LKB1/AMPK axis inhibited the growth of breast cancer cells [131]. Inflammation is a key activator for numerous oncogenic pathways [132]. Activation of inflammasome/IL1β promotes breast cancer angiogenesis and progression [133]. Likewise, It was shown that inflammasome and IL1β are involved in tumor growth and metastasis and IL1 receptor antagonist significantly inhibited tumor growth and metastasis in the mice model of lung cancer [134].
Here, it is understood that Sestrin2 is involved in the pathogenesis of several types of human cancer and Sestrin2 expression can be used to determine the prognosis of cancers. Furthermore, increased expression of Sestrin2 generally helps cancer chemotherapy and overcomes resistant tumors.
Oxidative stress
Oxidative stress, a disturbance in the balance between the production of reactive oxygen species (free radicals) and antioxidant defenses, is induced in cerebral ischemia especially through inflammation and reperfusion, which increases the production of ROS [111]. Xanthine oxidase and NADPH oxidase are the two key oxidative enzymes that play a major role in the production of superoxide anion, a key radical after stroke. Van Hemelrijck et al. using a rat model demonstrated that hydroxyl radicals (OH•) are elevated 2 h after stroke onset [112]. Nitric oxide (NO), a key radical, is produced by enzymatic conversion of L-arginine by three types of nitric oxide synthases (NOS) namely neuronal (nNOS), endothelial (eNOS), and inducible NOS (iNOS), which are elevated after brain ischemia. Although endothelial NOS is upregulated directly after brain ischemia, neuronal NOS and inducible NOS are upregulated only after a day and even later, respectively [112]. Nitric oxide has both beneficial and detrimental roles in cerebral ischemia. Although NO plays a major role in restoring blood supply to the ischemic area, reducing brain damage, it also reacts either with superoxide anion to form radicals or with free electrons to form peroxynitrite, contributing to lipid peroxidation, cellular toxicity, and eventually cell death [113]. In addition to the action of NO on free radicals, it also enhances the expression of adhesion molecules and inflammatory mediators, inhibits enzymes necessary for DNA replication, and promotes iron loss from cells [114]. The pathological effects of NO on brain tissue damage largely depend on the sensitivity of the cell to NO, concentration of NO, and whether the inflammatory phase is acute or chronic [115]. Knockout animal studies showed that mice lacking nNOS had reduced infarct volume, which demonstrates that NO produced by nNOS leads to tissue damage. Alternatively, injury after ischemic stroke increased in mice lacking eNOS showing the protective function of eNOS by dilating the blood vessels resulting in normal blood flow to the penumbra [116]. Furthermore, disruption of iNOS did not affect infarct volume after day 1, but after 72 h, iNOS disruption increased infarct size [117]. Various antioxidants such as vitamins, lipoic acid (LA), and N-acetylcysteine have been tested for efficacy in stroke [118]. Vitamin E and its analog MDL 74,722 have been reported to reduce lesion volume and behavioral impairments in rodent ischemic stroke models [119,120,121]. Alternatively, a follow-up study in human subjects demonstrated that vitamin E and C did not reduce the risk of ischemic stroke and did not enhance functional recovery in stroke patients, respectively [122,123,124]. In addition, other antioxidants such as EPC-K1, a phosphate diester of vitamin C, reduced lesion size, lipid peroxidation, and renal reperfusion injury in rat model [125, 126]. Apart from dietary antioxidants as supplements which had little or no effect in clinical trials, NXY-059, tirilazad, edaravone, and citicoline are being studied in clinical patients for its efficacy in treatment of ischemic stroke [118]. However, NXY-059 and tirilazad failed to provide clinical improvement in larger clinical trials [127, 128]. In 2001, edaravone was approved in japan to treat acute phase cerebral infarction. In 2015, edaravone was approved by Japan and in 2017, by the US Food and Drug Administration, for treatment of amyotropic lateral sclerosis [129]. However, a clinical study demonstrated that early-stage edaravone treatment delayed progression of infarction and edema and reduced acute-phase mortality, but edaravone alone did not cause any significant functional recovery [130]. Citicoline is now being reported as a potential therapeutic candidate for ischemic stroke. Citicoline, which is also called as cytidine 5’-diphosphocholine (CDP-Choline), is a combination of two molecules, cytidine, and choline. These molecules are efficient in crossing BBB and then they combine to form CDP-Choline in brain cells [68]. During ischemia, phosphatidylcholine is broken down into free radicals and fatty acids that worsen ischemic injury [131]. The hypothesized mechanism is that citicoline undergoes hydrolysis followed by dephosphorylation to form cytidine and choline and these two molecules act as substrates to re-form citicoline in the brain. This process minimize phospholipid breakdown and enhance phospholipid resynthesis for membrane repair [132]. In addition, citicoline also scavenges free radicals providing antioxidant and anti-inflammatory roles after ischemic damage [133]. Clark and his colleagues reported that 24 h post-MCAO, administration of citicoline for 28 days enhanced motor and functional recovery [131]. Due to promising biological properties, citicoline has also been administered (500 and 2000 mg, i.v.) in a randomized controlled trial [134]. However, other randomized, placebo, controlled clinical studies have reported that citicoline has limited benefits [135,136,137]. The controversial results might be due to methodological limitations such as odda ratios rather than risk ratio and the use of cumulative meta-analysis rather than trial sequential analysis. The results of International Citicoline Trial on Acute Stroke (ICTUS) was published by Davalos et al. where 2298 patients with moderate to severe stroke (within 24 h) of the anterior territory were randomly assigned to double-blinded treatment with 2000 mg citicoline daily or placebo for 6 weeks. Assessment for baseline characteristics and a known risk factor for stroke showed that citicoline-treated patients had well-balanced baseline characteristics risk factors for stroke. However, no significant difference was noted in primary outcome of recovery assessed by mRS score. Evaluating other pre-specified subgroups showed beneficial effects of citicoline in patients older than 70 years and in patients with less-to-moderate stroke. Thus, up-to-date meta-analysis of all clinical trials of citicoline resulted in overall beneficial effect with an odds ratio of 1.14 of achieving good clinical outcome compared to controls [138]. Despite the pros and cons, citicoline is the only promising drug in confirmative clinical trials and no other neuroprotective compound had any positive effect on subgroup analysis [138].
Cytokines
Cytokines are immunomodulating agents and they play a major role in cell activation, proliferation, and differentiation [139]. Cytokines are generally small pleiotropic polypeptides (8–26 kDa) barely detectable in the brain with their receptors constitutively expressed at very low levels. Cytokines play a major role in upregulating the expression of cell adhesion molecules (CAM) [140, 141]. Especially, intracellular adhesion molecule 1 (ICAM 1) upregulation in the ischemic core leads to BBB disruption by aiding recruitment of leukocytes, which in turn release cytokines. BBB disruption causes migration of various inflammatory cells such as macrophages, natural killer cells, T lymphocytes, and polymorphonuclear leukocytes to the ischemic site. Various studies have demonstrated that infiltrating leukocytes and microglia elevate cytokines and some studies have reported that resident neurons and glia also produce cytokines following brain ischemia [106, 142, 143]. However, upon brain injury, the expression of pro- and anti-inflammatory cytokines is upregulated, but their spatial and temporal upregulation largely depends on the type of ischemic model used [72, 144, 145]. The three major proinflammatory cytokines are interleukin 1β (IL-1β), tumor necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6) that provoke and aggravate an inflammatory response after stroke [146, 147]. Post-mortem studies demonstrate that TNF-α positive cells are observed in brains of severe ischemic stroke patients from 3 days post-stroke, staying positive until 15 months [148]. Tumor necrosis factor-α serum levels are increased within 6 h post-stroke and their levels are maintained for 10 days [149]. Similarly, IL-1β levels are elevated in the CSF with peak levels at days 2 and 3 post-stroke [150, 151]. However, some studies have shown no increase in IL-1β levels in serum and plasma, which might be due to IL-1β localization at the inflammatory site [152]. Interleukin-1β mediates ischemic, traumatic, and excitotoxic brain injury through its action on neurons, glia, and vasculature. Similar to TNF-α and IL-1β, Il-6 levels are elevated in CSF of severe stroke patients. Few studies report that CSF IL-6 levels increase within 24 h and peak at days 2 and 4 [153], while some studies report peaking at days 3 and 7 [154, 155]. However, their levels appear to be dependent on stroke type and severity. Interleukin 1β, an important mediator of neuroinflammation, upregulates the expression of IL-6. Hence, IL-1β receptor antagonist, anakinra administration, demonstrated good clinical improvement and decreased peripheral neutrophil count and IL-6 levels [156]. Alternatively, transforming growth factor-β (TGF-β) and IL-10 are anti-inflammatory cytokines that inhibit the expression of proinflammatory cytokines thereby reducing inflammation after ischemic stroke [157, 158]. These pro- and anti-inflammatory agents act as predictors and help in prognosis in ischemic injury. However, other cytokines also contribute to brain damage and repair, but the balance between the beneficial and detrimental effects of cytokines largely depends on the biochemical and physiological status of the brain [158].
Chemokines
Chemokines are small signaling proteins (8–10 kDa) belonging to the family of cytokines. They have the ability to induce directed chemotaxis in nearby responsive cells, especially leukocytes [159, 160]. There are 40 different chemokines known so far; they all share a similar structural pattern with 4 cysteine residues and, based on that, they are classified into four subfamilies that play a major role in stroke with C-X-C attracting neutrophils and C-C attracting monocytes/macrophages [161]. The other two classes being C and CX3C where Cs denote two N-terminal cysteine residues and depending on whether amino acid is between them or adjacent to them, they are classified as CXC and CC, respectively. Similar to cytokines, chemokines have both unique and overlapping receptors, which belong to the superfamily of G-protein-coupled receptors [162]. Identical to cytokines, chemokines and their receptors are usually expressed in low concentrations [163, 164], but after cerebral ischemia, TNF-α and IL-1β enhance the production and release of specific chemokines such as cytokine-induced neutrophil chemoattractant (CINC), monocyte chemoattractant 1 (MCP-1), microglial response factor-1 (MRF-1), and fractalkine and macrophage inflammatory protein 1 (MIP-1) which are upregulated in the first 3 h and remain elevated for at least 6 h [163]. Chemokines and their receptors play a major role in modulating various pathological and physiological processes, in which their role in post-ischemic inflammation is an important contributor to ischemic brain injury [165]. Brait et al. used PCR arrays to screen temporal expression profile of several chemokine-related genes using focal cerebral ischemia (occlusion for 30 min) in mice. Gene analysis at 4, 24, and 72 h reperfusion showed that several chemokines belonging to CXC family were upregulated (> 10-fold), mediated leukocyte infiltration and played a major role in stroke pathogenesis [166]. Since chemokines have been implicated in the worsening of stroke pathogenesis, their ligands and receptors act as potential therapeutic targets. One such chemokine, chemokine ligand 2 (CCL-2) and its receptor CCR2 signaling, mediates pathological post-ischemic inflammatory response by not only inducing leukocyte recruitment but also disrupts BBB and leukocyte adhesion to brain endothelial cells in an MCAO (45 min occlusion) mice model [167, 168]. CCL-2 levels increase in ischemic penumbra from 6 h of reperfusion with peak levels at 24 and 48 h [169, 170]. Levels of CCL2/CCR2 are positively correlated with infarct size and enlargement of the ischemic lesion. Moreover, CCL2 expression is upregulated in the CSF and serum in ischemic patients [171]. Thus, genetic deletion or manipulation of CCL2/CCR2 expression may be a therapeutic target for ischemic stroke. CCL2 gene disruption diminished infarct volume in focal cerebral ischemic mice model (30 min occlusion), and CCR2 deletion not only reduced infarct size and brain edema but also enhanced motor functions in focal transient cerebral ischemia mice model with 30 min occlusion [169, 172]. Alternatively, overexpression of CCL2 exacerbates ischemic injury in mice [170]. In a recent study using CCR2 knockout mice, after MCAO and reperfusion, the infarct size was less in CCR2 KO mice with lower mortality when compared to WT control when measured 3 days after stroke. Nonetheless, CCR2 KO mice had high mortality and neurological deficit from 5 to 28 days after stroke. Hence, CCR-2-dependent monocyte/macrophages not only aggravate brain injury but also alleviate functional recovery after ischemic stroke. Few reports demonstrated that CCR-2-dependent monocyte infiltration to the stroke-injured hemisphere peaked at 3 days after stroke, but after day 7, monocyte-derived macrophages (MDM) exhibited both proinflammatory and anti-inflammatory phenotype equally, but after 2 weeks, macrophages with anti-inflammatory phenotype dominated. However, blocking monocyte recruitment using anti-CCR2 antibody at 1 week post-stroke eliminates long-term behavioral recovery with significant decrease in anti-inflammatory gene expression in an MCAO mice model with 30 min occlusion [173]. Apart from the anti-inflammatory mechanisms, MDM regulate and control long-term and acute microglia-mediated neuroinflammation [174]. Hence, manipulation of periphery macrophage control of microglia could be a therapeutic option for treatment for microglia-mediated CNS diseases. Apart from proinflammatory properties of CCR2, CCR-2 recruit bone marrow-derived monocytes/macrophages to prevent hemorrhagic infarct transformation and these cells help maintaining neurovascular unit integrity following ischemia in both photothrombotic and tMCAO mice models [175]. Hence, pharmacological manipulation of CCR2 must be deliberately investigated to avoid impairment of normal physiological function of CCR2. Similar to CCL-2, macrophage inflammatory protein 3α (CCL20) has also been suggested to be involved in proinflammatory macrophage recruitment to the ischemic brain followed by IL-1β and TNF-α production [176, 177]. The initial production of chemokines is attributed to activated microglia (MIP-1α, MIP-2, and MRF-1) followed by astrocytes and injured neurons (fractalkine and MCP-1) after cerebral ischemia [72, 144, 178]. Under pathological conditions, such as brain injury, chemokines act as signals released into cerebrospinal fluid (CSF) and extracellular fluid to recruit neutrophils, monocytes, and microglia [179] whereas under normal physiological conditions, chemokines govern the positioning of cells in tissues and recruit leukocytes to the inflammatory site [180]. Leukocyte recruitment is achieved by chemokines working in harmony with adhesion molecules to affect BBB permeability through diapedesis (passage of blood cells through intact walls of capillaries, generally along with inflammation). Kim et al. also proved that mRNA expression of monocyte chemoattractant protein 1 (MCP-1) was nearly undetectable under normal physiological conditions but after ischemia caused either by permanent or temporary MCAO for around 12 h or 2 days, resulted in a significant increase in MCP-1 mRNA expression, which persisted for up to 5 days. Supporting this hypothesis, Chen et al. proved that overexpression of MCP-1 exacerbated ischemic brain injury (24–48 h following occlusion) along with enhanced infiltration of inflammatory cells in MCAO (2 h occlusion) mice model [181]. Similar to MCP-1, hindering the activation of MIP-3-α resulted in reduced infarct size in a transient MCAO rat model with 2 h occlusion [182]. Chemokine overexpression results in chronic neutrophil infiltration, persistent glial activation, and BBB disruption, resulting in terminal wasting syndrome, but on the other hand, chemokine knockout mice show deficiency in leukocyte recruitment [163, 183]. Apart from their chemotactic properties, chemokines directly affect BBB. Co-culture of brain endothelial cells and astrocytes showed that addition of MCP-1 resulted in a significant increase in BBB permeability and it causes alteration in tight junction proteins (TJP) in endothelial cells and the detrimental action of MCP-1 is diminished by the absence of chemokine receptor type 2 [184]. Hence, chemokines may be a potential target for therapeutic interventions.
Excitotoxicity
Ischemic stroke causes major ATP and phosphocreatinine depletion that results in the release of excitatory amino acids that leads to excitotoxic neuronal damage called excitotoxicity. Barone et al. reported that accumulation of potassium ions and acidosis are preceding events in the ischemic cascade leading to ionic disturbances [185]. Increase in potassium (K+) levels leads to the release of glutamate, which in turn stimulates Na+/Ca2+ channels coupled to N-methyl-D-aspartate receptors (NMDAR). This further elevates Na+ and Cl- levels along with passive influx of H2O resulting in cyotoxic edema. Extracellular glutamate also activates α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and metabotropic glutamate receptors, which is a critical step in the inflammatory cascade [186]. Metabotropic and NMDA receptors work through monoionic channels and incidentally enhance intracellular Ca2+ levels. Various detrimental pathways including voltage and receptor gated Ca2+ influx leads to a significant increase in free cytosolic calcium levels thereby creating mitochondrial calcium overload and further compromising ATP production [185]. Moreover, high intracellular Ca2+ levels lead to the activation of proteases, lipases, kinases, phosphatases, endonucleases, and free radicals that promote breakdown of phospholipids, proteins, and nucleic acids [187, 188]. In normal physiological state, Mg 2+ blocks channel pores of NMDA receptor, but when glutamate is released from pre-synaptic sites and when AMPA receptors are activated, Mg 2+ is completely removed from NMDARs due to partial depolarization in post-synaptic membrane. This causes influx of Na+ and CA+ into the cell that plays a major role in ischemic cell death [189, 190]. Calcium overload inside neurons activates a series of downstream death signaling pathways such as calpain activation [191], ROS production [192], and mitochondrial impairment [193]. Hence, NMDAR antagonists have been rigorously studied as a therapeutic candidate for treatment of ischemic stroke. In specific, GluN2A- and GluN2B-containing NMDAR are the two important NMDAR in adult forebrain. During an ischemic event, activation of synaptic and extra synaptic GluN2B-containing NMDARs leads to excitotoxicity followed by neuronal apoptosis. Alternatively, activation of GluN2A-containing NMDARs also results in neuroprotection and neuron survival [194, 195]. Hence, dual roles of NMDARs might depend on subcellular locations and subtypes of the receptors that are activated. GluN2B, PSD95, and nNOS complexes play a major role in the activation of death signaling pathways during ischemic stroke [196]. Post-synaptic density protein-95 (PSD-95), a scaffolding protein, links NMDARs to downstream molecules, such as nitric oxide synthase (NOS). PSD-95 is made up of three PDZ domains in which PDZ1 and PDZ2 bind to threnonine/serine-x-valine-COOH (T/SXV) motif at the intracellular c-termini of GluN2 containing NMDAR subunits [197]. Also, PDZ2 of PSD95 binds to N-terminus of nNOS. Both these events lead to Ca2+ influx from overactivation of nNOS, followed by excess production of nitric oxide (NO), an effector of excitotoxicity [198]. Hence, disrupting GluN2B-PSD95-nNOS complex impairs NO production and prevents excitotoxicity based neuronal damage [199]. One such study showed that “Tat-NR2B9c or NA-1,” an interfering peptide that binds to either PSD95 or nNOS, prevents the activation of downstream neurotoxic pathways and neuronal superoxide production in neuronal cells [200]. In vitro studies have also demonstrated that administration of Tat-NR2B9c after ischemic stroke reduced infarct volume and improved behavioral outcomes [201]. Cook and his colleagues mimicked clinically relevant situations in a gyrencephalic non-human primate MCAO model and demonstrated that Tat-NR2B9c reduced infarct size (MRI and histology), maintained the ability of ischemic cells to preserve gene-transcription in genome-wide screens, and also prevented behavioral impairment [199]. A double-blinded, randomized, controlled proof-of-concept study conducted across 14 hospitals in the USA showed that NA-1 administration decreased ischemic infarcts. This study was performed on patients who had ruptured or unruptured intracranial aneurysm amenable to endovascular repair because diffusion-weighted MRI showed 90% of patients undergoing endovascular repair show small, embolic procedurally induced ischemic stroke [202]. In addition to peptides, small molecules targeting GluN2B-PSD95-nNOS complex are being studied. In vitro and in vivo studies demonstrated that ZL006 was reported to selectively obstruct PSD95 and nNOS interaction during ischemia. In addition, ZL006 did not affect the normal physiological role of MNDARs and nNOS [196, 203]. Similarly, IC87201 disrupted pathogenic PSD95-nNOS interaction without impairing normal nNOS activity in neurons [204]. However, biochemical and biophysical studies using fluorescence polarization (FP), 1H-15N.HSQC NMR and isothermal titration calorimetry have shown that under applied in vitro conditions, both ZL006 and IC87201 do not interfere with PDZ domains of nNOS or PDS-95 and it also does not inhibit nNOS-PDZ/PSD-95-PDZ interface [205].
Currently, safety and optimal neuroprotection of Neu2000 in ischemic stroke with endovascular recanalization (SONIC) trial is being performed to evaluate the neuroprotective efficacy of Neu2000 before endovascular thrombectomy in ischemic stroke patients [206]. Neu2000, a sulfasalazine derivative, selectively blocks NMDA receptors along with robust free radical scavenging property. Preclinical animal models demonstrated favorable efficacy and therapeutic window profile [207]. Apart from these small molecules, neuroprotective efficacy of peroxynitrite scavenger, disufenton sodium (NXY-059), uric acid, and antioxidants (edaravone) were evaluated using rodent models and clinical trials (discussed in detail in the “Oxidative stress” section). Free radicals also promote BBB disruption, brain edema and it has been reported that there is a significant decrease in free radical scavenging enzymes (superoxide dismutase) and increase in NO levels during ischemic stroke. In conclusion, decrease or decline in cerebral blood flow drains energy that is required for cellular ionic homeostasis. Ischemia-induced depolarization results in increased glutamate release, which leads to activation of endonucleases [208]. Hence, NMDA and AMPA receptor antagonists can be developed as neuroprotective agents that can inhibit depolarization and prevent ionic perturbations. Glutamate receptor-mediated excitotoxicity is also activated by death molecules like Fas ligand, generated by matrix metalloproteinase, matrilysin [209]. Tissue inhibitor of matrix metalloproteinase 1 (TIMP1) inhibits excitotoxic-mediated neuronal death [210]. Hence, the role of MMPs in aggravating ischemic neuronal death is discussed below.
Matrix metalloproteinases
Matrix metalloproteinases (MMPs) are a large family of proteolytic enzymes that degrade all components of extracellular matrix [211]. MMPs range from matrilysin (267 amino acids), being the smallest member of the family to large transmembrane proteins such as MMP-14 (582 amino acids). All MMPs have a definitive configuration consisting of the catalytic zinc site, the propeptide region, the fibronectin binding site, and the transmembrane site. MMPs are broadly classified into constitutive (MMP-2 and MMP-14) and inducible (MMP-3 and MMP-9) enzymes, where constitutive enzymes act close to the site of activation whereas inducible enzymes are not constrained to act at the activation site [53]. Although, MMPs act as proinflammatory factor, they are also important for normal physiological function such as neuronal regeneration, cell proliferation, angiogenesis, and apoptosis [212]. MMPs play a major role in BBB disruption during the acute phase of ischemic stroke by degrading basal lamina and weakening the blood vessels. MMP-9, an inducible MMP, is a 92-kDa type IV-collagenase initially secreted in latent form and is activated by proteolytic processing in the extracellular space. Studies using ischemic rodent models demonstrated that there is a significant increase in the expression of pro/active MMP-9 within 24 h following ischemia in rats [213], and they have been detected in both central and peripheral cells with a unique expression profile [214, 215]. MMP-9 along with tissue plasminogen activator (tPA) has been reported to disrupt BBB resulting in hemorrhagic transformation [216]. Rosenberg et al. using an MCAO rat model with 2 h occlusion demonstrated that the activity of MMP-9 was maximally elevated at 48 h [217]. MMP-9 is initially produced by endothelial cells and neutrophils, and after 5 days, they are produced by macrophages. During ischemic stroke, endothelial cells overexpress MMP-9 within and at the periphery of ischemic lesions that results in increased vascular permeability. Type IV-collagen, laminin, and fibronectin are the key components of basal lamina that separates cerebral blood vessels from extracellular matrix. Overexpression of MMP-2 and MMP-9 can digest basal lamina, and this digestion begins as early as 2 h following ischemia, which corresponds with BBB breakdown 3 h following ischemia [218]. Moreover, following stroke, alterations in other MMPs such as enhanced levels of pro/active MMP-2 [219], MMP-3 [84], MMP-10 [219], and MMP-13 [220] have been reported. MMPs have been associated with increase in circulating cytokines [221] and escalation of thrombolysis [222] along with activation of microglia and astrocytes [84]. Various studies demonstrated that MMP inhibition not only reduces infarct size but also alleviates brain edema and hemorrhage [53, 223]. Moreover, when compared to wild-type mice, MMP-9 knockout animals had smaller infarct. Whereas, a similar effect was not observed in MMP-2 knockout mice which demonstrates that MMP-2 may be involved with neovascularization whereas MMP-9 may be involved in edema [224]. Apart from this, MMP levels in plasma could be developed as potential biomarkers since they can be used to predict the severity of stroke. First-ever ischemic stroke patients enrolled in intensive rehabilitation study demonstrated that MMP levels were stable as healthy volunteers during the study period but baseline MMP-12 and MMP-13 were correlated with stroke severity. Surprisingly, plasma MMP3 was significantly increased in patients with better motor/functional recovery [225]. However, serum level of MMP-9 was independently positively correlated with initial stroke severity, as well as with clinical recovery [226]. Since MMPs play both beneficial and detrimental roles in stroke, MMPs are explored as potential therapeutic targets that are reviewed in detail by Yang et al. [53]. Further, MMP-mediated BBB permeability in ischemic stroke is inhibited by cyclooxygenase 2 inhibitors [227]. Hence, modulating the activity of Cox-2 or prostaglandin E2 prevents MMP-mediated BBB disruption.
Cyclooxygenase—an arachidonic acid metabolite
Activation of immune cells results in the release of phospholipase A2 (PLA2) that leads to initiation of the arachidonic acid (AA) cascade by hydrolyzing glycerophospholipids. This results in subsequent energy failure and depletion of ion concentrations due to intracellular calcium accumulation [228]. Tabuchi et al. demonstrated that AA metabolites act as signaling molecules that initiate a post-ischemic immune response in MCAO (75 min occlusion) mice model. Further, PLA2 deficient mice had small infarcts when compared to wild-type mice demonstrating their detrimental role in brain ischemia [229]. Cyclooxygenase (COX) further metabolizes AA to prostaglandin H2, once released from brain phospholipids. Cox is present in three isoforms namely Cox-1, Cox-2, and Cox-3 [230], where Cox-1 is constitutive and Cox-2 is inducible. Twenty-four and 96 h following brain ischemia by MCA occlusion, Cox-1−/− mice had larger infarcts when compared to COX-1+/+ mice which might be due to severe cerebral blood flow reduction in the vulnerable region at the periphery of the ischemic territory [231]. Thus, vascular function of COX-1 plays an important role in maintaining cerebral blood flow in post-ischemic brain. Alternatively, pharmacological inhibition of Cox-1 using Valeryl Salicylate in a model of global cerebral ischemia with 5 min occlusion increased the number of healthy neurons in the hippocampal CA1 even after 7 days post-ischemia [232]. Under normal conditions, Cox-2 is involved in synaptic plasticity and cerebrovascular regulation [233, 234]. Whereas, under disease conditions, their reaction products have a major role in glutamate excitotoxicity [235]. Cox-2, an essential isoform for prostanoid synthesis, has been reported to be enhanced within the ischemic border zone in rat models of focal cerebral ischemia [236]. Autopsy studies have also demonstrated that Cox-2 immunoreactivity has been observed in vascular cells, infiltrating neutrophils and also in neurons sited at the border of an infarct in stroke patients [237, 238]. Prostanoids, a sub-class of eicosanoids, which is reported to be a key factor in the pathological mechanism of ischemic and excitotoxic brain injury, is derived from Cox-2 [235]. Long-term treatment with Cox-2 inhibitors has been shown to elevate the incidence of myocardial infarction and stroke [239]. Hence, it is mandatory to specifically target downstream effectors of Cox-2. Specifically, Cox-2-mediated neurotoxicity is achieved through prostaglandin E2, a downstream effector molecule that acts through four G-protein-coupled receptor namely EP1, EP2, EP3, and EP4 [240, 241]. These receptors have distinct signal transduction profiles and mostly opposite cellular actions [241]. Various studies support the detrimental role of EP1 subtype of prostaglandin E2 receptor in cerebral ischemia [240, 242]. Moreover, administration of EP1 receptor antagonist ONO-8713 followed by striatal unilateral NMDA injection prevents neurotoxicity and diminishes ischemic and excitotoxic brain injury [242, 243]. EP1 receptors augment neurotoxicity by impairing Na+–Ca2+ exchange leading to disruption of Ca2+ homeostasis and resultant excitotoxic neuronal death. Moreover, pharmacological inhibition of EP1 receptor with SC51089 (EP1 antagonist) 6 h after MCA reduced brain injury suggesting their importance for therapeutic development [240].
Transcriptional modifications
It is well known that following cerebral ischemia, there is upregulation of mitogen-activated protein kinase (MAPK) and nuclear factor kappa beta (NF-κβ) gene expression, which both play a key role in activation of inflammatory signals [244]. NF-κβ family shares a Rel homology, and this heteromeric transcription factor is usually made up of a sequel of Rel subunits such as Rel (cRel), Rel A (p65), Rel B, NF-κβ1 (p50 and its precursor p105), and NF-κβ. The most common composition of NF-κβ is p50 and p65 and is normally found in the cytoplasm bound to its inhibitor protein IκB. IκB kinase (IKK) phosphorylates an inhibitor of kappa B (IκB) that leads to ubiquitination and dissociation of IκB from NF-κβ and eventual degradation of IκB by the proteosome. This process helps NF-κβ to translocate to the nucleus and bind to specific sites of DNA and in promoter domains of proinflammatory genes that lead to transcription of TNF, ICAM-1, COX-2, iNOS, and IL-6. There is a strong correlation between oxidative stress-mediated neurotoxicity and elevated NF-κβ expression. NF-κβ expression contributes to the increase in cell death after MCAO and its activation results in enhanced expression of downstream target genes that play a vital role in neuronal injury. Moreover, either inhibition of p50 or p50 in knockout mice models protects from brain ischemia [245, 246]. Further, Hermann et al. reported that inhibition of IkK markedly reduced infarct size and in contrast activation of IkK enlarged the infarct size. This work is also supplemented by a selective small molecule IkK inhibitor studies that mimicked their genetic studies [247]. Activation of NF-κβ and MAPK pathways also leads to the expression and activation of nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) pyrin domain containing 1 and 3 (NLRP1 and NLRP3) inflammasome protein that contributes to neuronal cell death and brain injury following 1 h occlusion in a MCAO mice model [248].
Mitogen-activated protein kinase (MAPK)
Mitogen-activated protein kinase (MAPK) family is composed of three groups namely extracellular signal-regulated kinase ½ (ERK ½), c-Jun N-terminal kinases (JNK), and p38 [249]. Various stress factors such as cytokines, osmotic stress, and microtubule disorganization stimulate MAPK pathway that leads to activation of three-tiered Raf/MEK/ERK cascade through G-protein-coupled receptors. Stress activated protein kinases (SAPK), JNK, p38 MAPK, and ERK have been reported to play a detrimental role in brain ischemia [250]. Following brain ischemia, activation of MAPK pathway was noticed to occur 30 min and 3 days, and moreover, many proinflammatory mRNA transcriptions are mediated by p38 MAPK that suggests its role in inflammation-mediated ischemic brain injury. Following ischemia (90 min occlusion), p38 MAPK signaling plays a major role in ischemia-induced astrogliosis, while p38 inhibition attenuated hypoxia and scratch injury-induced astrogliosis in a MCAO mice model [251]. Phosphorylated p38 MAPK was detected in astroglia [252], microglia [253], and neurons [254] of ischemic brain tissue that demonstrates its role in the inflammatory response. Increased inflammatory factors can strongly activate P38 MAPK forming an injury cycle [255]. Following 2-h middle cerebral artery occlusion, MAP Kinase/ERK pathway plays a major role in the expression of MMP leading to BBB breakdown and upregulation of proinflammatory factors [256]. Also, inhibition of the MAPK cascade via suppression of cytokines through anti-inflammatory drugs, which blocks p38 MAPK, arrested the production of TNF-α and IL-1β resulting in neuroprotection [185]. Moreover, treatment with a neuronal membrane lipid precursor (CDP-Choline) after ischemic stroke resulted in the decrease of phosphorylation in ERK 1/2, MEK ½, and ElK-1 transcription factors [19]. Hence, there is increasing evidence that MAPK is a significant regulator of ischemic damage that leads to the possibility of using MAPK as a therapeutic target. Moreover, dioscin, a natural steroid saponin, decreased MAPK phosphorylation and inhibited HMGB1 translocation to the cytosol that resulted in less proinflammatory response [257]. MEK/ERK inhibitor U0126 also decreased HMGB1 expression in reactive astrocytes [258]. These reports suggest that MAPK and HMGB1 pathway act as an important factor in stroke pathology.
High-mobility group box protein family
Alarmins or danger-associated molecular patterns (DAMPs) are released by dying or necrotic cells, initiating an inflammatory response in ischemic core. DAMPs released in the blood stream also help recruit peripheral immune cells. Several DAMP-s such as nucleic acids, ATP, S100 proteins, and HMGB1 have been found to contribute to the inflammatory response in stroke [259]. High-mobility group box (HMGB) proteins are ubiquitous and abundant DNA binding proteins, and they can act as a trigger of neuroinflammation.A unique structure of HMGB1 helps them to bind to DNA and intracellular proteins to mediate DNA repair and transcription [260, 261]. HMGB1, also known as amphoterin, was initially described as a non-histone DNA binding protein with high electrophoretic mobility. It plays a major role in nucleosomal structure stabilization and binds to the minor groove of linear DNA without sequence specificity resulting in association of nucleoprotein complexes and recruitment of transcription factors. Although HMGB1 plays a major role in the conservation of nuclear homeostasis, it also acts as an extracellular signaling factor involved in cell proliferation, differentiation, and pathogenesis [262]. Various studies reported that HMGB1 is released in the brain after cytokine stimulation and is associated with inflammation [263, 264]. During cellular stress such as stroke, HMGB1 functions as a proinflammatory cytokine [265, 266]. Following cerebral ischemia in mice, HMGB1 translocates from nucleus to the cytoplasm or within 1 h, it disappears from the cells completely [259]. The expression levels of HMGB1 in microglia, astrocyte, and blood vessels increased dramatically 2 h after MCAO in mice [267]. Chin et al. recently demonstrated that HMGB1 protects oligodendrocytes and prevents white matter injury during ischemic stress. Mice injected with glycyrrhizin, a specific inhibitor of HMGB1, resulted in expansion of demyelinating lesion along with exacerbated sensorimotor behavioral deficits [268]. HMGB1 is also released by dying oligodendrocyte to act as an autocrine factor under ischemic condition [269]. Clinically, HMGB1 were found to be significantly higher in serum or plasma of patients with ischemic stroke when compared to age- and sex-matched controls [270]. The expression levels of TLR2 on monocytes either stimulated with or without HMGB1 were evaluated in ischemic stroke patients. Real-time PCR and ELISA assays resulted in higher expression of TLR2 in monocytes of stroke patients stimulated with HMGB1. Anti-TLR2 immunomodulation diminished the expression of IL-17, IL-6, and IL-33 [271]. Faraco et al. reported that HMGB1 promotes induction of iNOS, COX-2, IL-1β, and TNF-α and also increases excitotoxic as well as ischemic neuronal death in vitro [272]. In addition, there is a strong correlation between MMP-9 and HMGB1 levels in ischemic stroke patients, which was associated with poor outcome [273]. Toll-like receptors 2 and 4 and receptor for advanced glycated end products (RAGE) can bind to extracellular HMGB1 and active transcriptional factor NF-κB [274,275,276]. MyD88, a downstream effector molecule of TLR signaling has been shown to be involved in HMGB1-mediated post-ischemic inflammatory response and enhances stroke lesions when compared to MyD88 knockout mice in fMCAO model [277]. Downregulation of HMGB1 by RNAi (RNA interference) resulted in less microglial activation and reduced infarct volume in rodent MCAO models [278, 279]. Various studies have demonstrated that blocking or modulating HMGB1 by compounds such as statins (atorvastatin, fluvastatin) and by shRNA provided neuroprotective effects against ischemic stroke [280,281,282].
Hence, similar to other inflammatory mediators, HMGB1 plays an important role in aggravating detrimental events during ischemic stroke. Table 2 summarizes the beneficial and detrimental role of key inflammatory mediators associated with stroke.
Table 2 Beneficial and detrimental role of inflammatory factors associated with ischemic stroke
Full size table
To conclude, a cascade of these neuroinflammatory events leads to activation apoptosis and resultant cell death. The molecular events initiated in the brain after ischemic stroke involves a cascade of intracellular mechanisms such as failure of ionic pumps, activation of glutamate receptors that leads to excitotoxicity, increase in calcium influx, and enhanced ROS release that leads to DNA damage and mitochondrial impairment. Intracellular calcium influx is enhanced through activation of N-methyl-D-aspartate (NMDA), D,L-α-amino-3-hydroxy-5-methyl-isoxazolpropionic acid (AMPA) glutamate receptors, or through acid-sensing ion channels (ASICs). Cerebral ischemia leads to increased ROS production and activation of Fas death receptors which results in the activation of pro-apoptotic caspase-8. Increased ROS results in DNA damage and phosphorylation of p53 that activates nuclear cell death pathways. Caspase-8 or calpains activation further leads to cleavage of Bid to truncated Bid (tBid). Truncated Bid fuses with Bax, which in a normal physiological condition, is neutralized by B cell leukemia/lymphoma 2 (Bcl-2) or Bcl-xL. In ischemic state, tBid and Bax interaction leads to mitochondrial membrane depolarization and resultant release of cytochrome C or apoptosis-inducing factor (AIF) into the cytosol. These events initiate caspase-dependent or caspase-independent neuronal death. Released cytochrome C interacts with pro-caspase 9 and apoptotic protein activating factor-1 (Apaf-1) to form apoptosome that leads to the activation of executor caspases such as caspase-3. In addition, AIF transloacates to the nucleus causing DNA fragmentation and resultant cell death. Further, cell death cascade aggravates due to release of damage associated molecular patterns (DAMPs) by damaged neurons that result in activation of microglia, astrocyte, and endothelial cells and inflammasome formation. These events further orchestrate release of cytokines, ROS, and BBB disruption. In addition, BBB disruption causes influx of peripheral immune cells that exacerbates inflammatory pathways (Fig. 3). Numerous studies both in pre-clinical and clinical platforms are being performed in search of novel therapeutic strategies to either inhibit or slow down pathological mechanisms associated with ischemic stroke (Fig. 4). Though various neuroprotective studies have resulted in frustrating clinical trials, their results also provided us with knowledge on understanding the mechanisms involved in ischemic cascades. However, currently, there remains a pressing need for research and development of stroke therapies that can be successfully replicated in clinical trials for prevention as well as for early critical care in stroke patients.
Fig. 3
Apoptotic mechanisms involved in ischemic cell death
Full size image
Fig. 4
Neuropathological mechanisms in ischemic stroke and respective targets assessed in clinical trials with and without beneficial effects
Full size image